
中金:新药研发过程经历了什么?
本文来自微信公众号“中金点睛”,作者:邹朋,赵利建等。
国内目前处于创新药的黄金发展期,但新药研发耗时长、投入大、风险高,其研发过程目前还不为大家所熟知,因此我们对新药发现的发展历史进行了回顾,梳理了目前新药研发经历的整个过程,以期对投资者了解创新药的研发过程和创新药投资提供帮助。
新药研发的技术变革
新药研发的历史也是一部新药研发技术变革的历史,新的技术手段的出现不断带动着新药研发行业的发展,新药研发技术的历史变革大致可以分为以下几个阶段。
19世纪以前,药物多来自于自然界的天然产物
在早期,人们主要根据自身经验利用天然植物、动物、矿物直接用于部分疾病的治疗,如毒性、催吐、止泻、止痛等效应。18世纪后,化学工业得到发展,人们开始分离、提取、纯化天然动植物的有效成份作为药物:1804年左右,Friedrich从罂粟中分离出吗啡,发现可以用于镇痛;1820年,Joseph和Pierre从金鸡纳树皮中分离得到了奎宁,可用于治疗疟疾;1831年,Heinrich从颠茄类植物中分离出了阿托品,具有散瞳效应;1885年,Nagai从蛇麻黄中分离出麻黄素,当时主要用于扩瞳。
20世纪前20年,合成药物开始出现
随着药理学和有机化学等科学的发现,人们可以在实验室里人工合成一些既有的药物,并合成一些自然界不存在的全新化合物。1859年,Gilm合成了纯的乙酰水杨酸(阿司匹林),但当时其药理作用并未被发现,1897年拜耳在实验室中合成了乙酰水杨酸,1899年作为解热镇痛药进行推广;1903年,Hermann和Joseph人工合成了巴比妥,拜耳将其作为镇静催眠药推向市场;1905年,Alfred 合成了普鲁卡因,其安全性高于可卡因,并且没有成瘾性,很快被推向市场;1906年,Ehrlich带领团队合成了大量有机砷化合物,经过筛选之后发现其中砷凡纳明(606)可以选择性杀死梅毒螺旋杆菌。
20世纪30至50年代,大量新药涌现
早期药理学主要以动物为研究对象,20世纪初期之后,以人体为研究对象,研究药物在体内的代谢、作用机制的现代药理学逐渐发展完善,加快了药物疗效确证和筛选的速度,新药研发行业也取得了很快的进展。抗生素、维生素、磺胺类药物、精神病药物、麻醉镇痛、疫苗等领域都出现了很多新药。
抗生素:1928年,Fleming发现了青霉菌分泌的青霉素具有很强的杀菌作用,但当时由于提取的青霉素不纯,临床应用有限,1941年生产工艺和提取方法的改进以及二战的需求,使得青霉素得到了广泛应用。此后人们又从成千上万的土壤样本中寻找到了链霉素、金霉素、土霉素、四环素、氯霉素、头孢菌素、万古霉素等抗生素。
维生素:1914年美国公共卫生部门开始研究糙皮病的病因,其发现摄入鸡蛋和牛奶后发病会减少,后来在志愿者和患者的饮食试验中都证明了本病与饮食有关,1935年这种物质被提纯出来,即维生素B3(烟酸)。人和大鼠缺乏维生素A时,眼球容易干燥,1914年Elmer和Marguerite通过把黄油皂化得到了脂溶性物质命名为“因子A”,1947年两位荷兰化学家合成了维生素A。1907年,为了研究脚气病的病因,Axel用豚鼠建立了动物模型,豚鼠发生了坏血病的症状,而引用新鲜蔬菜和水果后症状会消失,命名为“水溶性因子C”,1930年Norman分析出了这一物质的结构,从而可以合成维生素C。同期其他维生素也陆续被发现和合成。
磺胺类药物:1932年,拜耳希望以606(砷凡纳明)的原理为基础发现能够杀死细菌的化合物,其合成了数多种化合物,用从患者身上得到的细菌建立了小白鼠的疾病模型,结果发现百浪多息对链球菌感染非常有效,并申报了专利;1956年巴斯德研究所发现百浪多息的作用原理是其能在体内分解出磺胺,而磺胺的结构在1909年就被公开了,于是大家纷纷开发磺胺相关的衍生化合物,众多磺胺类药物被迅速开发出来推向市场。
精神病药物:1947年普朗克实验室在研究抗组胺药时发现了异丙嗪具有镇静和抗组胺作用,当时被用于手术合并用药以减少麻醉剂的使用,1952年异丙嗪被探索用于精神分裂症的临床试验,对躁狂患者有很好的效果,1954年其被FDA批准上市。1949年,CIBA公司发现了利血平具有降血压的效果,但同时具有抑郁的副作用,1952年,Nathan经过临床试验发现利血平对缓解精神分裂症患者的症状非常有效,1954年利血平作为精神分裂药开始推广使用。1954年,罗氏开始进行对氯丙嗪的me-too药物研究,其合成了几十个化合物,后发现苯氮卓有镇静、抗焦虑作用,但催眠作用较弱,1966年上市后迅速成为重磅炸弹品种。
麻醉镇痛药物:1937年,Etto合成了哌替啶(杜冷丁),比吗啡有更强的镇痛作用。1938年Max和Gustav合成了美沙酮,具有镇痛和镇静作用,且成瘾性小。1943年,Nils等合成了利多卡因有很好的麻醉效果,由于其起效迅速、持续时间长,很快获得了成功。
降糖药:1929年,Karl通过兔子模型研究缩二胍类化合物的降糖作用,发现二甲双胍作用最强,但当时很快被胰岛素的研究热潮所淹没,1957年Sterne开始从事双胍类化合物降血糖的研究,证实了二甲双胍具有优异的降血糖效果。1958年苯乙双胍在临床试验中也显示了较好的降糖效果,但有乳酸中毒的不良反应。临床实践中,患者在服用磺胺药氨磺丁脲后出现血糖降低,1954年这种现象在动物试验和糖尿病患者中都得到了证实,1955-1966年众多第一代磺脲类降糖药被用于临床。
此外,1954年抗凝血药华法林上市;1960年第一个口服避孕药异炔诺酮上市;也都是这个时期的重要成果。
20世纪60-80年代,药物分子理性设计时期
随着生理、生化等医学基础学科的发展,一些与疾病相关的酶、激素、神经递质的受体和底物等被发现;物质分析检测技术如液相色谱、核磁共振谱等得到应用;以及计算机、信息技术的发展应用;开始出现了基于结构的理性分子设计,新药发现从以前的随机筛选发展为结构修饰,以及根据受体结构进行QSAR(定量构效关系)设计,新药研发的效率得到提高(但仍有一些新药是通过随机筛选或偶然发现的)。此阶段发现了β受体阻断剂、抗组胺药、血管紧张素转换酶抑制剂、抗病毒等领域的新药。
β受体阻断剂:1958年ICI开始基于药效结构研究药物,其发现如果可以找到可以与肾上腺素竞争结合心脏的受体,那么就可以缓解心绞痛的症状,1965年第一个明确的受体拮抗剂普萘洛尔上市,用于心绞痛、高血压的治疗,由于其发现过程应用了药物受体构效设计的理念,被认为是新药发现的革命性概念之一。此后阿替洛尔、普拉洛芬、美托洛尔等β受体阻断剂相继上市。
抗组胺药:1964年人们已经认识到组胺可以刺激分泌胃酸,但是传统的抗组胺药对胃酸分泌影响不大,这使人们意识到组胺受体可能存在不同的分型,而SK&F公司的研究证实了2型组胺受体的的存在。基于组胺的结构,SK&F公司设计合成了数百种2型组胺受体的拮抗剂,而后经过优化,不断改善活性和安全性得到西咪替丁,西咪替丁于1976年在英国上市,是第一个年销售额达到10亿美元的药物。西咪替丁的研发过程将药物发现方法从搜索药物变为设计药物,也是新药研发的重大里程碑。
抗病毒药:1950s晚期William合成了一个嘧啶类似物(碘脱氧尿苷),该化合物可以与尿嘧啶竞争参与遗传物质的合成,从而抑制肿瘤的生长,但结果显示其抗肿瘤作用并不明显,而有抗病毒活性,其于1962年上市是第一个抗病毒药物。1964年Jerome在胸腺嘧啶的结构上进行改进得到齐多夫定,可以与胸腺嘧啶竞争抑制核酸的合成,阻断病毒复制,成为第一个被批准治疗艾滋病的药物。1979年,Burroughs Wellcome合成了环磷酸腺苷类似物,经过优化后得到阿昔洛韦,对疱疹类病毒有较好的效果。此后利巴韦林等抗病毒药也逐渐被发现。
血管紧张素转换酶抑制剂(ACEI):60年代,肾素-血管紧张素-醛固酮系统的作用逐渐被研究清楚,于是众多公司开始寻找这一系统中的靶点进行降压药的研究,Squibb公司开始寻找ACE的抑制剂,1973年其发现与ACE底物类似的化合物也可以与ACE活性部位结合但不会激活酶,可以取得较好的抑制效果,后经过优化得到了卡托普利,此后又有依那普利等数十个普利类药物上市。
20世纪80年代后,基于靶点的新药发现逐渐展开
基因组学、蛋白质组学、生物信息学等现代分子生物学科得到发展,人们对生命和生理、病理的认知得到进一步加深;也出现了生物芯片、组合化学、虚拟设计、高通量高内涵筛选等新技术;以靶点为基础的新药研发模式得到应用,其不仅加快了对化学药物研发的进程,也开发出了单抗、基因治疗药物及基因治疗药物等。
神经氨酸酶抑制剂:神经氨酸酶位于病毒颗粒表面,对病毒从宿主细胞的释放必不可少。1987年CSIRO确证了流感病毒神经氨酸酶的三维结构,1989年Biota公司通过合理药物设计开发了神经氨酸酶抑制剂DANA,以此为基础通过对比神经氨酸酶的结构,进行特定结构修饰,得到了扎那米韦(吸入剂)。1992年Gilead尝试口服神经氨酸酶抑制剂的开发,经过计算机辅助设计,得到了一个活性很高的化合物,但同样无法通过胃肠道吸收;后续经过修饰,降低极性,得到新的化合物,其容易通过消化道吸收,而后在体内代谢为前述化合物而发挥作用,此新化合物即为奥司他韦。
伊马替尼:1960年人们发现慢性粒细胞白血病(CML)患者白细胞有一种短小的染色体(染色体易位造成),称之为费城染色体,1985年人们又发现费城染色体易位生成的高活性络氨酸激酶(BCR-ABL)是引起CML的原因,CIBA公司和Oregon健康与科学大学合作通过高通量筛选技术寻找络氨酸激酶的抑制剂,最终经过修饰改造得到了伊马替尼。
瑞林类药物:1971年Andrzej分离了促性腺激素释放激素(GnRH),以其结构为基础合成其类似物,也可以与垂体上的受体结合,当以非生理性频率长期大剂量使用时,垂体敏感性下降GnRH受体表达下降,导致促黄体生成素(FSH)、促卵泡生成素分泌下降,因此可以用于对性激素反应的癌症,如前列腺癌、乳腺癌、子宫内膜异位症瘤以及性早熟等。80年代早期,各制药公司开始对GnRH进行修饰改造,1985年Takeda与Abbott合作研发的亮丙瑞林在美国上市,此后Sanofi研发出布舍瑞林、阿斯利康研发出戈舍瑞林、Debiopharm研发出曲普瑞林等,以及瑞林类药物的缓释制剂逐渐被开发上市。
DPP-4类降糖药:胰高血糖素(GLP-1)是一种肠促胰岛素,可以促进体内胰岛素的分泌,但是GLP-1很快会被体内的二肽酶讲解,于是研发二肽酶抑制剂是降糖药的一个研发方向。1967年人们发现抑制二肽酶-4(DPP-4)后可以促进肠促胰素的分泌,于是相应的DPP-4抑制剂在80年代被发现,1995年诺华公司应用虚拟筛选技术得到了维格列汀,MSD通过筛选技术得到了西格列汀,BMS开发出了沙格列汀。
生物药:此阶段生物药的开发也得到快速发展,1988年诺和诺德推出了重组人胰岛素,大约同期重组人干扰素也得以上市。单抗技术也日臻完善,1998年开发出了以TNF-α为靶点的英夫利昔单抗和依那西普(融合蛋白),同年以HER2为靶点的曲妥珠单抗也得以上市,2004年以EGFR为靶点的西妥昔单抗上市,同年以VEGFR为靶点的贝伐珠单抗上市等。2017年两款CAR-T药物被FDA批准用于血液肿瘤的治疗,同年FDA批准基因治疗药物Luxturna用于RPE65突变的视网膜疾病的治疗。此外RNA药物和基因编辑等技术也到了快速发展。
新药研发过程[1]
新药研发过程是指从实验室发现活性化合物后不断优化改进和评价直至发展成为安全有效的药物的系统工程,其包含了发现和开发两大阶段。发现阶段主要包括作用机理的研究、大量化合物合成、活性化合物筛选、化合物优化到候选药物的过程,发现活性化合物的方法可以是理性设计(rational molecular design)、随机筛选,也可以是偶然发现。开发阶段是对候选药物进行临床前评价和临床试验评价的过程,需要对候选药物的药学、药理、毒理、安全性、有效性进行系统的评价。
图表:新药研发过程

注:上述为现在常用的基于靶点的新药研发过程,除此外随机筛选、偶然发现等也是重要的药物发现途径
资料来源:Nature Reviews,中金公司研究部
靶点发现
药物作用的靶点是指能与药物结合并产生药理效应的生物大分子,机体的病理发生过程是由多个环节构成的,当某个环节或靶点被抑制或激活,则可以达到治疗疾病的目的。靶点的类型主要有受体、酶、离子通道、核酸等。目前已上市药物的靶点仍以蛋白类靶点为主,其中受体占多数,且大多数分布在细胞膜上。
图表:药物作用靶点分布
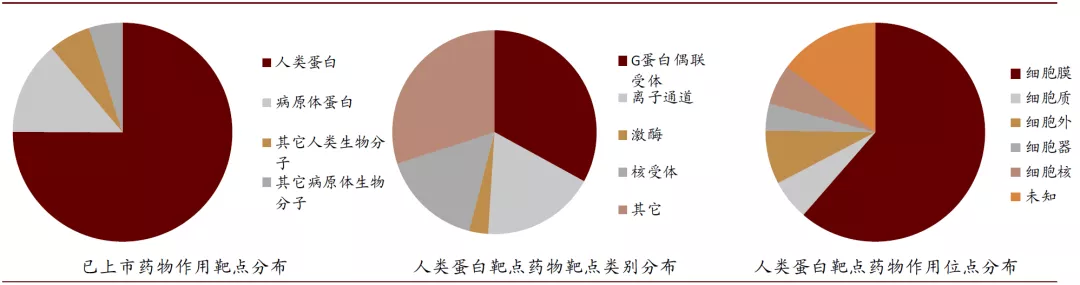
资料来源:Nature reviews, Nature biotechnology,中金公司研究部
80年代以后,随着基因组学、蛋白质组学、等分子生物学科的完善,以靶点为核心的药物研发模式逐渐建立。当新的基因或蛋白质的功能被揭示之后,其分子生物学机制得到明确,就可以寻找该生物效应传导机制上的相关环节作为靶点,如受体常在生理效应中发挥信号转导的作用,针对其可以研发激动剂或拮抗剂,以激活或抑制相应的生理活动。
基因组学和蛋白质组学研究成为靶点发现的重要途径。
在基因水平上:通过寻找正常状态和病理状态下的基因表达和基因序列差异的分析,可以找到疾病相关的基因;通过疾病相关基因的功能分析,包括基因敲除、过量表达、RNA干扰、反义mRNA技术等可以对该靶点进行确证。
在蛋白质水平上:通过比较正常状态和病理状态下蛋白表达的差异,可以找到相关疾病的靶点,常用的技术包括凝胶电泳、质谱分析、蛋白质芯片、同位素亲和标签等;此外通过蛋白质相互作用也可以为新药研发提供靶点,常用的方法有酵母双杂交、噬菌体展示、双分子荧光互补等。
靶点评价。不是所有的靶点都会被应用,因为部分靶点可能位于细胞内甚至细胞器内导致药物难以到达,或者其所关联的疾病已经有了更好的治疗靶点,再或者它们对应市场难以激发商业上的开发兴趣。对靶点的评价通常从以下几方面进行:
靶点确证:是否有遗传和化学证据表明靶点对细胞的生长、存活有重要意义;
可药性:是否对对靶标体系的机制有较为清晰的理解,相关抑制剂/激活剂的结构是否具有类似药物的物化特性;
检测技术:是否已有较为成熟的分析检测手段;
结构信息:是否已有高解析度的蛋白-配基复合体结构信息。
模型建立
选定靶点之后,需要建立生物学模型以筛选和评价化合物的活性。建立模型可以在不同层次,如体外分子(受体、酶等)水平、组织细胞水平、离体器官、动物模型体内试验等。
动物模型:1980年以前多数药物的评价都建立在动物模型之上,这类方法首先在实验动物上建造一种模型,模拟人体的疾病,并且用临床有效的药物证明该模型是有效,进而用来评价化合物的活性。这种评价体系虽然耗费的时间和费用都比较大、且速度较慢,但是整体动物试验可以评价的内容丰富,除了化合物活性外,对药效学、药代动力学、和安全性都有一定程度的评价,因此得到的化合物结构可靠,成功概率高。至今,这种方式仍在应用。
离体靶点分子模型:在以靶点为核心的药物发现模式中,由于已经证实某些蛋白质与疾病相关联,因此可以用基因工程的方法克隆、分离和纯化得到相应的离体靶点蛋白,在体外评价化合物对该靶点蛋白的作用。伴随着基因组学、高通量筛选、组合化学等技术的发展,这种方法得到了广泛应用,因为其具有快速、可以同时对大量化合物进行评价的优点。但实际中也有人认为,用靶点分子来表征复杂的疾病系统过于简单化,一些疾病的发生是多因素导致的,由于化合物对离体分子发挥作用从而期待其可以治疗相应疾病的过程显得过于跳跃。
组织细胞模型:该方法是将病理状态下的组织或细胞与正常组织或细胞相比较,证实该功能的缺陷对疾病的发生具有特异性,然后建立细胞或者组织模型评价化合物对生物功能的影响。近年发展起来的高内涵筛选(high-content screening)、细胞系统生物学等技术是与此类模型相契合的。其并非针对单一靶点的筛选方法,但也并没有使用整体动物,而是将生物学过程综合到细胞内,可以在单一试验中对同时检测化合物对细胞生长、分化、凋亡、代谢途径以及信号转导的影响,从而确定化合物的生物活性和潜在毒性。
苗头化合物[2]
苗头化合物是对特定靶点具有初步活性的化合物,苗头化合物是新药研发的物质准备的起点。其可以通过多种途径得到,如化合物库的随机筛选、天然活性物质的筛选,以及基于靶点结构的理性药物分子设计、虚拟筛选等。
随机筛选:该方法是发现苗头化合物最常用的方法,是从化合物库中对所有化合物的作用进行评价以筛选出有活性的化合物。为了保证苗头化合物的质量和入选率,化合物库的结构应该多样化、数量较大、具有良好的类药性。目前大型制药企业均建立了自己的化合物库。
天然活性物质筛选:天然活性物质通常是根据传统医药或民间用药的经验有目的的筛选得到的,天然药物在新药研发中也占有非常重要的地位,临床应用药物的40%是天然产物或半合成的天然产物。动物、植物、微生物和海洋生物的物种多样性和生存环境的多样性决定了其代谢产物具有多样性,其可以视为天然的化合物库。
合理药物设计:合理药物设计是根据靶点的三维空间构型,并参考内原活性分子和外源活性分子的化学结构设计出针对该靶点的化合物。合理药物设计具有目的明确、设计出的分子质量更具有合理性、减少所筛选的化合物数量以及缩短研究开发周期等优点。
虚拟筛选:虚拟筛选是借助计算机和专业软件的从大量化合物中挑选出一些苗头化合物,进行活性评价。实体的药物筛选需要建立大规模的化合物库,并提取或培养出大量的靶点蛋白或者细胞,因此需要的投入较大。而虚拟筛选是讲筛选过程在计算机上模拟,可以节约经费开支,常用的方式有基于分子对接的虚拟筛选、基于药效团的虚拟筛选、基于定量构效关系的虚拟筛选、基于药代动力学的虚拟筛选。
基于片段的药物发现(Fragment-based lead discovery,FBDD):高通量筛选虽然能够获得高活性的化合物,但是其往往忽略分子的成药性,使得其可能因为药代性质等缺陷而无法成药。而化合物与靶点的结合可以看作其分子部分片段与靶点的结合,基于片段的筛选方法是通过对片段化合物库的筛选发现活性片段,由于片段的分子结构简单,1)片段化合物库的数量较小,FBDD可以通过筛选较少数量的片段化合物探索更大的化学空间;2)结构简单使得片段分子更易与靶点结合,发现苗头分子的几率更高;3)片段分子通常拥有更高的配体效率(结合自由能与分子中重原子数比值),后续优化设计更加容易。2011年FDA批准了首个FBDD的药物,Vemurafenib用于晚期黑色素瘤的治疗。
目前,如核磁共振、X射线衍射、质谱等等都已应用到FBDD当中。核磁共振可以测定片段分子与靶标蛋白的结合强度以及结合位点,通过结合强度归纳片段分子的构效关系,通过结合位点分析能够指导片段分子进行进一步的设计与优化。在设计片段化合物库的时候,Congreve在Lipinski的“5原则”基础上提出了片段分子的“3原则”:1)片段分子的相对分子质量小于300;2)脂水分配系数小于3;3)氢键的给体和受体数量均小于3。
图表:基于片段的药物发现的原理
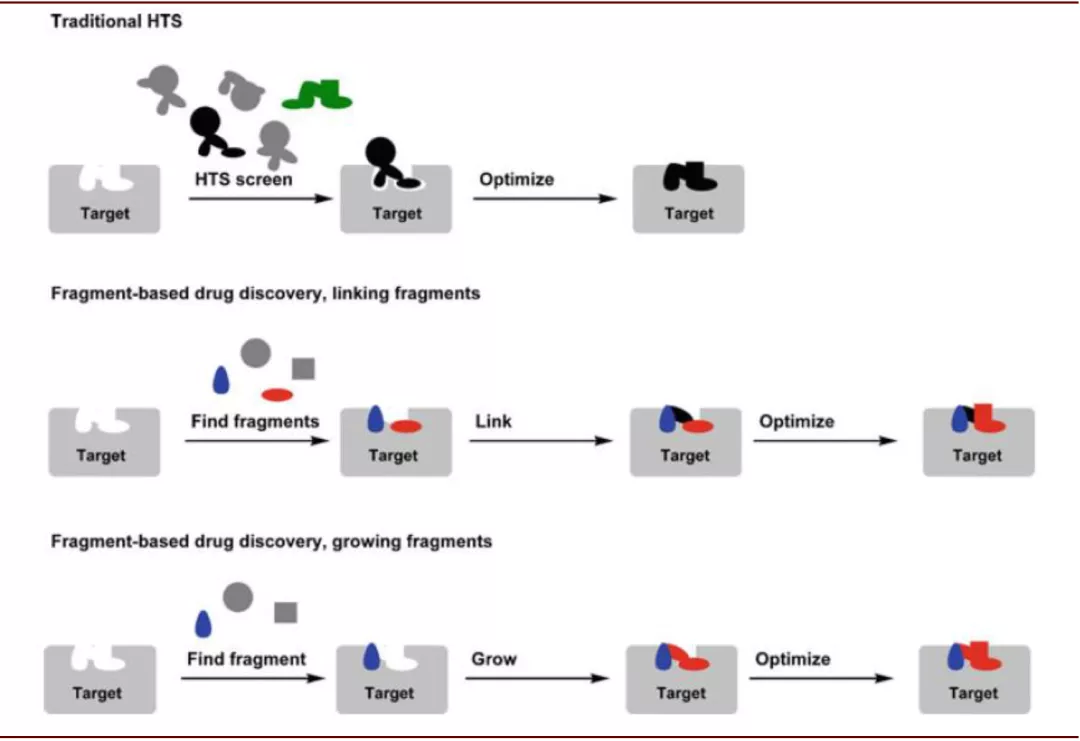
资料来源:Springer,中金公司研究部
苗头化合物评价标准。以上方法筛选出的化合物并不一定都适合作为苗头化合物,苗头化合物应基本满足如下的标准:1)与靶标的结合强度不低于10μmol/L;2)有一定水溶性,溶解度不低于10μg/mL;3)可穿越细胞膜;4)细胞水平显示生物活性;5)无细胞毒作用;6)具有化学稳定性;7)可以制备获得;8)具有知识产权的保护[3]。此外Lipinski也根据大量临床口服药物的化学结构,归纳出一些经验性特征成为遴选苗头化合物、先导化合物和构建化合物库的重要参考标准,即口服药物的类药5原则:1)相对分子质量低于500;2)氢键给体低于5;3)氢键接受体低于10;4)clogP(脂水分配系数)低于5。
先导化合物
苗头化合物是对特定靶点具有初步活性的化合物,与先导物还有一定差距。先导化合物是具有特定生物活性的化学结构,其或许存在活性不强、选择性较低、吸收性较差、毒性较大等缺陷,但通过对其进行修饰改造,可能得到具有良好药理作用的候选药物。
苗头化合物向先导化合物过渡。苗头化合物向先导化合物演化没有固定的程式,主要取决于苗头化合物的质量和对靶点信息的认知。如果已知靶点的三维结构,以及与配体形成复合物的结构,则可基于靶点结构进行分子模拟,从微观结构上优化苗头化合物。如果没有靶点和配体的结构信息,可用苗头化合物的拓扑结构进行分析,例如将线性化合物逐段进行变化,将环状化合物分区进行变换,以探索不同结构变换对活性的影响。常用的方法有骨架的优势结构变换、骨架迁越、建立构效关系进行药效团的探索、简化结构或者调整极性等。
发现先导化合物的途径。先导化合物与苗头化合物并没有绝对的界限,以上用来发现苗头化合物的方法也都可以用来发现先导化合物,除以上方式外,先导化合物的发现还有以下途径:
组合化学与高通量筛选:传统化学合成的方式是一次合成一个化合物进行评价,而组合化学则可以在短时间内合成大量的化合物形成化合物库,其具体做法为将一系列结构、反应性能接近的化合物构成一个模块(假设其中含有n种化合物),与另一构建模块的m种化合物进行反应即可得到n×m种化合物,再将之与另一构建模块的p种化合物反应即可得到n×m×p种化合物,随着反应步数的增加,化合物数量将以几何级数增加。因此组合化学方法可以同时制备含大量分子的化合物库,而将之与高通量筛选技术结合,可极大的加快先导化合物发现和优化的速度。
图表:组合化学原理
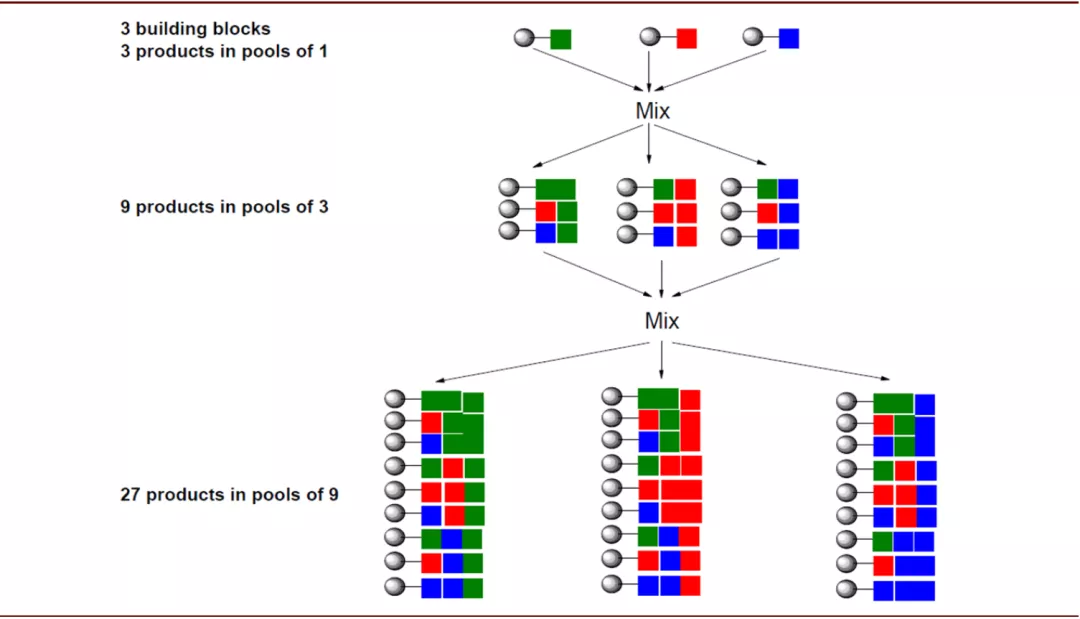
资料来源:University of Zurich,中金公司研究部
DNA编码化合物库(DNA-Encoded Compound Libraries,DEL):DEL技术的建库原理也是基于组合化学技术,但在建库的过程中每个分子结构都对应一段DNA序列,最终合成的每个化合物都有一段独特的DNA序列与之相链接, 化合物的合成完成后将所有化合物与靶点蛋白进行相互作用,去除与靶点不结合或结合力较弱的化合物,将结合力强的化合物所带的DNA序列进行PCR扩增和测序即可知道相应化合物的结构,从而筛选出先导化合物。与高通量筛选相比,DEL在化合物数量、筛选周期及成本方面拥有较大的优势,DEL技术下几十亿化合物库已经司空见惯,最新的DEL库化合物数量可达40万亿。
图表:DNA编码化合物库的构建过程
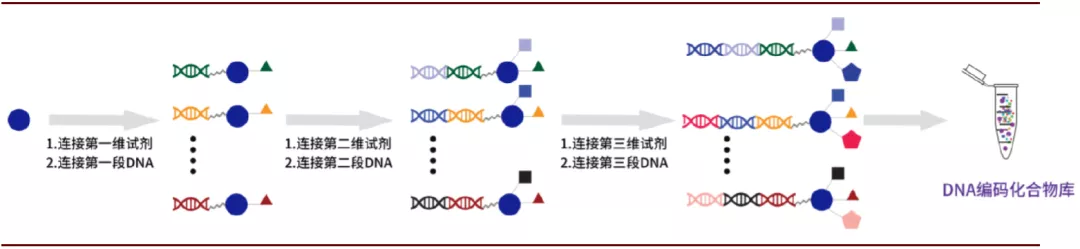
资料来源:成都先导招股书,中金公司研究部
图表:DNA编码化合物库的筛选过程
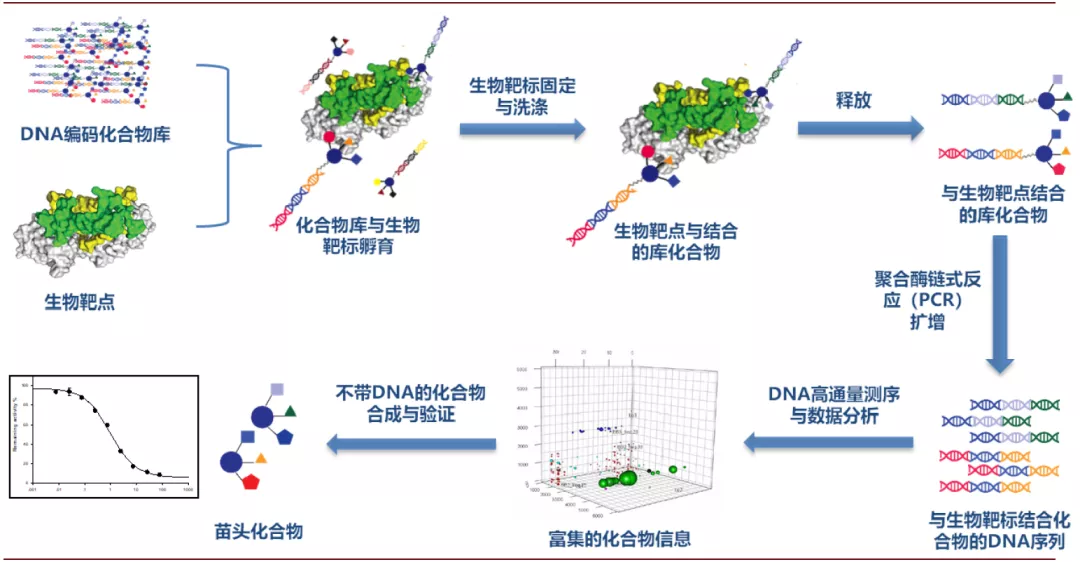
资料来源:成都先导招股书,中金公司研究部
计算机辅助设计:计算机辅助设计是在靶点功能、靶点空间结构、内源性配基的化学结构、靶点受点与配基的结合性质等信息的基础上,借助计算机自动构造出结构和性质与受点互补的配基分子的三维结构,通常按照空间互补和静电互补作用相互关系等来设计分子。其可以在既有的三维结构数据库中,逐一的寻找符合特定性质的结构;也可以设计全新的药物分子,根据受点的形状和性质,利用计算机程序直接设计新的配基分子三维结构。[4]
基于配体或底物的分子设计:在靶点三维结构未知的情况下,也可以通过分析对同一靶点具有活性的生物活性分子的结构,得到三维构效关系模型,通过计算机推测靶点的空间构型,以此虚拟靶点的三维结构进行药物设计。称之为基于配体/底物结构的药物设计,具体方法包括3D-QSAR、药效团模型法等。[5]
基于代谢作用:药物进入体内后,会在肝脏内经过I相、II相代谢反应,转化成有利于排出体外的水溶性代谢产物,药物经过代谢之后一般会失去或降低活性,但也有部分药物经过代谢之后活性反而提高,这类代谢物也可以作为先导化合物。如解热镇痛药非那西丁在体内经过代谢后会转化为对乙酰氨基酚,对乙酰氨基酚的解热阵痛强度强于非那西丁,现在已经作为常用的解热阵痛药物。
基于药物的副作用:理想的药物是只对特定靶点发挥作用,这是药物的特异性,但是人体内存在大量的具有多种生理功能的生物大分子,如受体和酶,药物很难只对唯一的靶点发挥作用(药物的杂泛性),因此药物会在临床上产生不良反应。但有时观察临床副作用,研究其作用机制,也是发现新药的线索,通过将副作用发展成主要作用,摒弃原有的主要药理作用,也是发现先导化合物的重要途径,即为“老药新用”,如黄酰脲类的降血糖作用被开发为降糖药、西地那非在心血管疾病的临床试验中副作用被开发为治疗ED的药物。
偶然发现:以上讨论的方法多数是有目的的合成和筛选,但生物体是非常复杂的体系,而自然界物质的多样性有时候为发现药物提供了偶然的机会,如青霉素的发现、苯二氮䓬类抗焦虑药等。
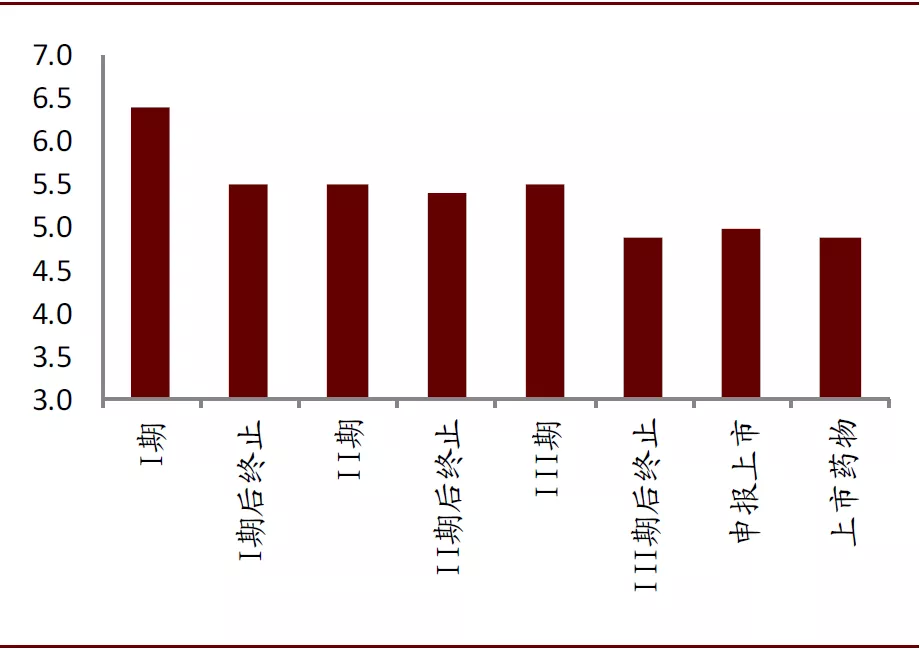
活性不是苗头、先导化合物的唯一评价标准。筛选苗头化合物、先导化合物不能仅以活性强度作为单一指标而忽视其他因素。但相对分子质量、亲脂性、水溶解性等是影响药物药代的重要因素,忽视这些因素易导致后续开发遇到难题,例如在苗头化合物的后续优化中人们往往加入基团或者片段,以增加与靶点结合的机会,提高活性强度,但相对分子质量过大会导致药物不易吸收、过膜性差、部分基团容易被代谢等。Wenlock对不同研发阶段药物的相对分子质量进行了统计,上市药物的相对分子质量要明显低于在研药物,氢键接受体也有类似的现象。
图表:不同研发阶段药物相对分子质量

资料来源:American Chemical Society,中金公司研究部
图表:不同研发阶段药物氢键接受体数量
资料来源:American Chemical Society,中金公司研究部
先导化合物的评价标准。先导化合物的质量直接影响新药研发的速度和成败。先导化合物也没有统一的评判标准,且不同类别的药物评判标准不尽相同。但从经验来看其在药效、药代动力学、理化性质方面应达到一定要求,而且能够获得专利以保护研发药物的知识产权。
图表:先导化合物的参数标准
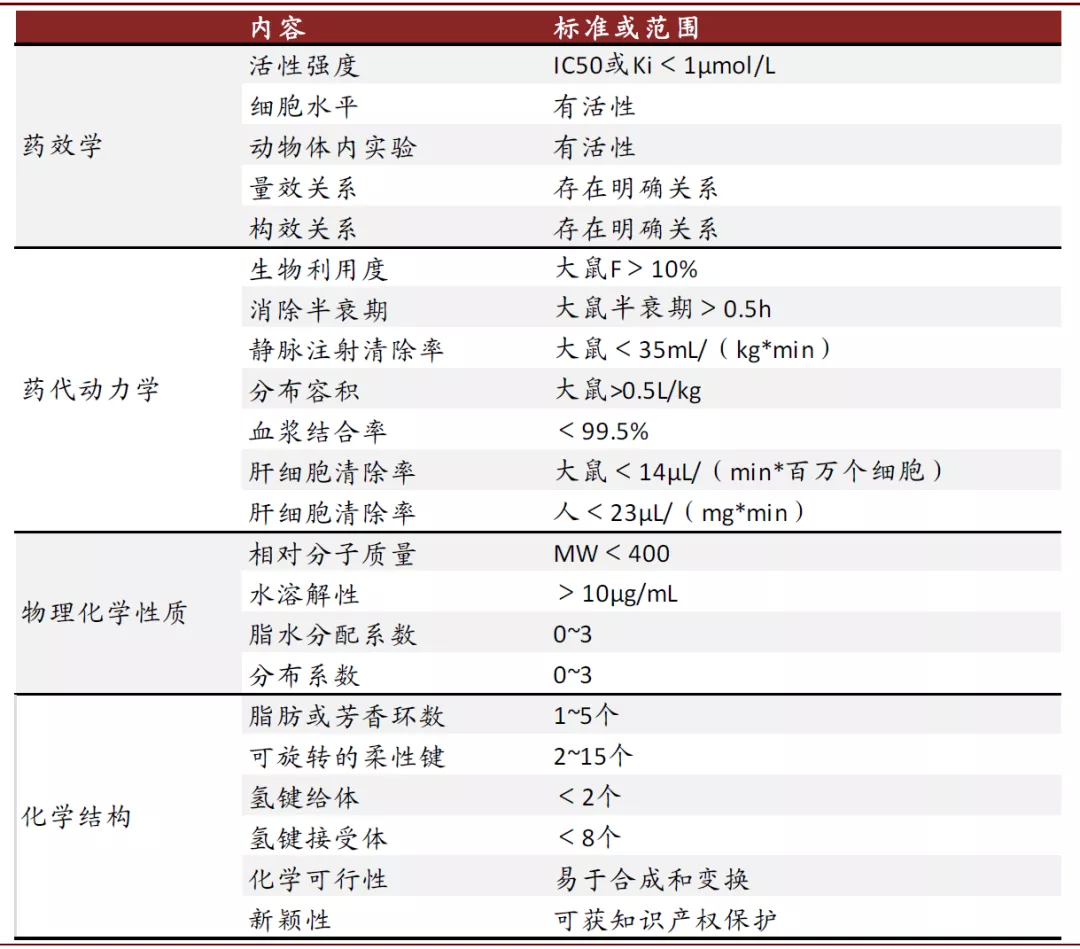
资料来源:药物设计策略,中金公司研究部
候选药物[6]
先导化合物往往存在一定的缺陷,如活性强度不够、选择性不高、药代动力学不合理、理化性质不适宜等,因此不能直接作为药物,需要对其结构进行变换和修饰,进一步优化后才能成为候选药物。优化先导化合物的方法通常有剖裂和剪切、拼合、局部修饰、生物电子等排、立体异构及外消旋转换等。
剖裂和剪切(Dissection & Shearing):剖裂和剪切是对比较复杂的先导化合物进行结构简化的操作,一般将先导化合物剖析成两个或数个片段,再将简化的片段进行活性评价和构效关系分析,提取出决定活性的基本结构和特征。例如对吗啡进行剖裂和剪切得到了右丙氧芬和美沙酮等副作用更低的阿片受体激动剂。
拼合(Association):与剖裂相反,拼合是将两个药物或其药效团经共价键结合成单一分子的操作,品和得到的药物成为栾药,拼合的目的是提高药物的作用强度,或同时作用于与疾病相关的两个不同靶点而提高疗效。
局部修饰(Local manipulation):局部修饰是先导化合物优化更常用的方法,局部修饰遵循了优化中“结构改变最小原则”,以渐进的方式避免活性的突然消失。局部修饰的内容包括:增加或减少饱和碳原子的同系化合物、链状化合物闭环或环状化合物开环、引入双键或手性中心、引入或除去或取代有空间障碍的大体积基团、改变基团的电性等。
生物电子等排(isosterism):电子等排是指具有相同数目的原子数和电子,且电子排列状况也相同的分子、原子、或基团具有相似的性质;生物电子等排是指分子或基团的外层电子相似、或电子密度有相似的分布、且分子形状和大小相似的结构可以产生大致相似或相关或相反的生物活性。通过生物电子等排基团的置换可实现对先导化合物的优化。
骨架结构变换(scaffold modification):骨架结构变换是基于药效团理论,其认为药物于靶标发挥作用并非整个分子都直接参与复合物的结合,而是少数原子或基团起主要作用,这些与靶点结合部分发生互补性结合的关键性原子和基团被称为药效团。药效团的特征可以分为正电中心、负电中心、氢键给予体、氢键接受体、疏水中心和芳环质心,不同药效团特征的组合和距离形成特定的药效团。而药效团是附着在化学骨架之上,通过化学骨架的变换可以保持药效团特征不变,但可以改善药物的药代性质。常用的方法有上述的电子等排,也有优势结构(优势结构是指可以衍生出对多种受体具有高亲和作用的分子骨架,是已有药物中常见的分子结构片段),以及骨架迁越等。
前药修饰(prodrug):部分通过体外筛选得到的化合物其生物利用度可能较低,或者由于其官能团的极性使之吸收性较差或者在体内分布不当,或者其存在首过效应导致在体内被过早代谢,需要对其进行修饰以改善其药代动力学性质。前药是一类在体外活性较小而在体内经酶或非酶作用后释放出生物活性物质,从而发挥药理作用。利用前药设计原理可以改善药物在体内的吸收、分布、转运和代谢等药代动力学性质。例如依那普利拉立体活性较强但消化道吸收较差,而制成的前药依那普利在人体胃肠道吸收较好,吸收后在肝脏被酯酶水解以原药形式发挥作用。
旋光异构体及外消旋转换(stereomeric drug):若先导化合物中含有一个以上的手性中心,则其会存在对映异构体;而由于体内靶点结合或代谢的选择性,有时一种对映体具有药理活性而另一种药理活性较弱、或没有药理活性、甚至有毒性。例如左旋氨氯地平具有较好的降压效果,而右旋氨氯地平几乎没有降压效果;沙利度胺左旋和右旋异构体都有镇静作用,但是左旋异构体会干扰孕妇的胎儿发育,导致了著名的“反应停”事件。
图表: 丙氨酸的旋光异构体
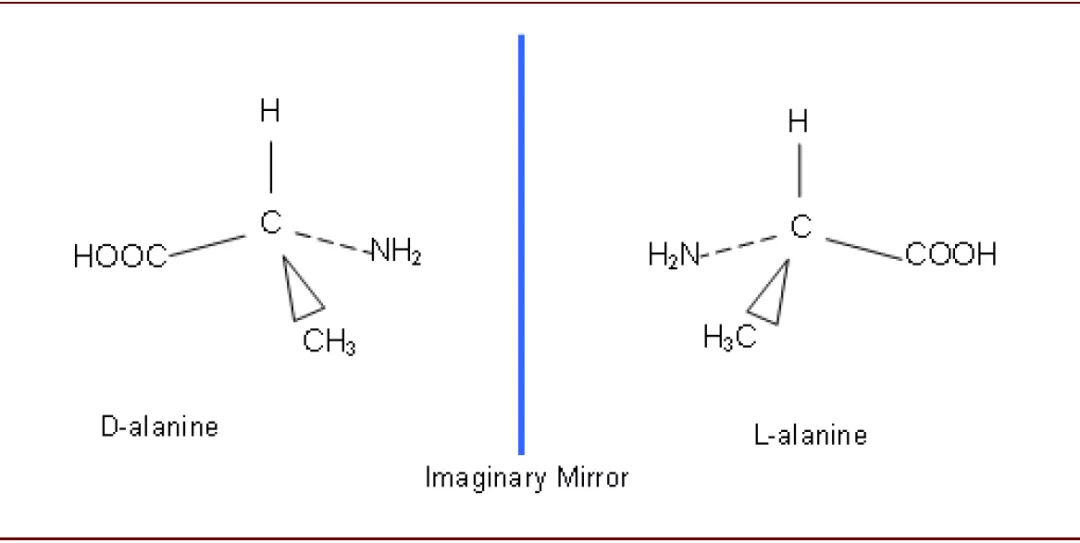
资料来源:chemguide,中金公司研究部
候选药物的确定。候选药物的确定标志着分子设计、化学合成、生物评价循环反馈的完成,达到了新药开发的标准。候选药物的确定也没有统一的标准,每个项目选择的候选药物数量也不尽相同,总的趋势是要求候选药物具有较好的成药性,为了降低研发失败的概率、缩短开发时间,候选药物的选定一般要遵循以下原则。
图表: 候选药物的确定遵循的一般原则
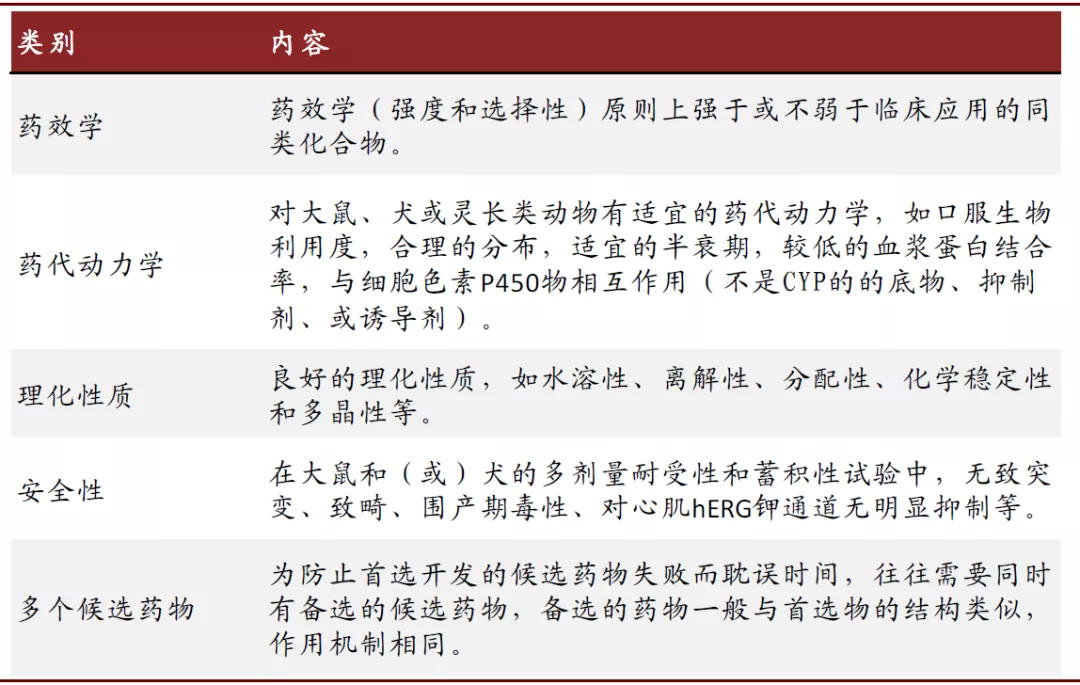
资料来源:药物化学总论,中金公司研究部
新的药物研发路线。除以上经典的药物研发技术路线之外,近期也出现一些更加创新性的新药研发路线,如PROTAC、RNA药物、基因编辑等。
临床前开发[7]
确定了候选药物之后,即可开展新药开发研究,对候选药物进行规范、系统的评价,评价过程必须符合GLP、GCP、GMP的规范。新药开发可分为临床前和临床研究两个阶段,临床前开发主要对候选药物进行人体外或动物体内安全、有效性评价以及工艺质量研究,从而为临床研究提供临床候选药物,是药物进入临床试验阶段不可或缺的一步。
图表: 新药临床开发过程
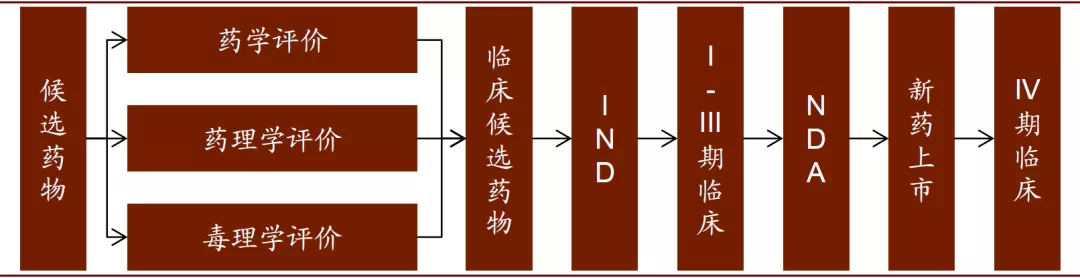
资料来源:CNKI,中金公司研究部
药学评价
药学评价是新药评价的基础,与新药的临床疗效和安全性密切相关,且贯穿了新药评价的全过程。主要评价内容有药物名称、结构、理化性质、合成路线和工艺、制剂处方和制备工艺、定性鉴别、含量测定、稳定性试验、质量标准的研究以及为临床前评价和临床试验提供所需的药品等。按评价对象可以分为对原料药的药学评价、对制剂的药学评价、及质量标准的研究和制定等。
原料药的药学研究:原料药的药学研究包括制备工艺研究、结构确证、命名、理化性质研究等。
制备工艺研究:包括试制路线、反应条件、合成工艺、原料来源和质量、中间体来源和质量、产品精制过程和工艺条件等。首先对拟合成的化合物进行文献调研,了解该化合物国内外研究情况、知识产权状况等,设计几条合理的合成路线,其中应该考虑起始原料获得的难易程度、合成步骤的长短、收率的高低、反应条件是否符合工业生产、环保要求等。起始原料药的选择应该质量稳定、可控,对起始原料引入的杂质应有一定的了解,必要时根据合成工艺的要求建立内控标准。在合成过程中对每一步工艺操作都应该有详细的描述,对工艺条件如反应装置、温度、压力、时间、溶剂、pH、光照等均应严格控制。
工艺优化与中试放大:原料药的制备工艺研究是在药物研发过程中不断优化的过程,通过反复的试验,结合原材料获得的难易程度、工艺路线的反应条件、环保和安全、产品的纯化等对生产工艺进行优化,以获得可行、稳定、收率较高、成本合理且是和工业化生产的工艺。在工艺优化过程中,应对重要的变化,如起始原料、试剂的种类或规格、重要的反应条件、产品的精制方法等发生改变前后对产品质量的影响,以及可能引入新的杂质情况进行研究。临床研究中应对中试或工业化生产规模的多批样品进行质量研究工作。
结构确证:原料药的结构确证是药物研发的基础,主要任务是确认所制备的原料药的结构是否正确,证明评价的新药是所预测的结构。同时需要对原料药的纯度也要进行一定的控制,供试样品的纯度应该大于99.0%。
命名:药品命名按照国家药典委员会的《中国药品通用名称命名原则》进行确定和申报,原料药的英文名应尽量采用WHO的International Nonproprietary Names for Pharmaceutical Substances(INN),中文通用名尽量与英文名相对应,一般以音译为主,也可以采取意译或意合译。药品制剂的命名,一般原料药名称列前,剂型名列后。
理化性质:对原料药的性状(外观、色泽、嗅、味、结晶形状、粒度、吸湿性、挥发性)通过观察和相应的方法测定,对其物理常数(溶解度、熔点、凝点、馏程、相对密度)按照药典规定的方法测定,对其油水分配系数、解离度(pKa)、晶型、立体异构现象也都要进行研究。
制剂的药学研究:药物必须制成适宜的剂型才能用于临床,药物剂型和制剂的设计需要遵循最大限度地发挥药效和降低不良反应的基本原则。制剂研究的目的是保证剂型选择的依据充分、处方合理、工艺稳定、生产过程能得到有效控制、适合工业化生产等。
剂型选择:结合临床用药目的和药物的理化性质确定药物适合的剂型,用于出血、休克等急救治疗的药物应选择注射剂型,用于呼吸道疾病急性发作应选择吸入剂;在胃液中不稳定的药物不宜开发为胃溶制剂,存在明显首过效应的药物一般考虑非口服给药途径的剂型。
处方研究:辅料的理化性质(性状、粒度、黏度、水分、pH值等)的变化会影响制剂的质量,处方研究应根据药物的理化性质、稳定性试验、药物吸收情况,结合所选剂型的特点选择适宜的辅料(辅料的选择应不与主要发生不良相互作用,不影响制剂的含量测定等)初步制定几种处方,再根据制剂基本性能评价(如溶出度、含量均匀度、pH值等)和稳定性评价筛选出初步确定的处方,但制剂处方的合理性最终还需要根据临床研究的结果进行判定,而在研究过程中发现对制剂质量、稳定性有影响的因素需要进行控制。
制剂工艺研究:根据剂型特点、药物理化性质等,设计几种基本合理的制剂工艺;分析并确定生产工艺过程中影响制剂质量的关键工艺、辅料和工艺参数,确立能够保证制剂产品质量的关键辅料用量和工艺参数范围。工艺过程需保证可重复,一般需要至少连续生产三批合格制剂产品。工艺放大是实验室制备技术向工业化生产转移的必要阶段,需要根据二者生产条件的差异,进一步优化工艺条件,确定适合工业化生产的设备和生产方法。
质量标准的制定:质量标准的制定应先确定质量研究的内容,然后进行方法学研究,最后确定质量标准的项目及限度。质量研究的内容应尽可能全面,根据原料药和制剂的特性、所采用的制备工艺、结合稳定性研究结果等制定,需要包含药品的结构、含量、理化性质、鉴别、纯度、杂质检查及限度控制、是否有多晶性、是否有旋光异构体,以及制剂的含量均匀度、溶出度、释放度、脆碎度、pH值等。
药理学评价
临床前药理学评价包括药效学、药代动力学和作用机制研究等。对该药物是否有效、量效关系、时效关系以及在生物体内的代谢变化规律进行研究,一方面为临床用药方案提供参考,也为新药申报临床提供依据。
药效学研究:临床前药效学应在理解疾病的发病机理和治疗措施的基础上,选择合适的体外和体内动物模型,合理的设计试验方案,以反映出药物是否有效、量效关系等。
试验对象:新药的主要药效作用应在用体内和体外两种以上试验方法进行证明,其中至少一种是整体的正常动物或模型动物。动物的选择应尽可能与人在生物学上接近、相应功能发达或敏感性高的动物。
试验设计:试验应设置空白对照和标准阳性物质对照,分组应随机进行。
观测指标:药物的疗效应靠客观的指标反映,能反映药效学的作用,如生理生化的化验指标、病理切片、X射线检查等,能半定量或定量。
剂量选择:应反映出量效关系,尽量求出半数有有效量(ED50)或有效剂量范围。
药代动力学研究:临床前药代动力学研究是通过人体外或动物体内试验获得药物的药代动力学参数,了解其在体内的动态变化规律,阐明ADME过程的动态变化规律和特点,从而为药效、毒理、临床研究提供参考资料。
试验对象:一般选取健康和成年的动物,常用的动物有小鼠、大鼠、兔、豚鼠、犬、小型猪和猴等,首选与药效学和毒理学研究一致的动物。对于创新性药物应选取至少两种动物,其中一种为啮齿类动物,另一种为非啮齿类动物;其它药物可选用一种动物,首选非啮齿类动物。
剂量选择:应至少设置3个剂量组,剂量的选择可参考药效学和毒理学研究所使用的剂量,最高剂量最好接近最小中毒剂量,中、小剂量根据动物有效剂量的上下限范围选取。
样品分析方法:常用的样品分析方法有色谱(气相色谱、高效液相色谱、色谱质谱联用等)、放射免疫分析、酶免疫分析、放射性核素标记法等。
药-时曲线:1)动物数量的确定:以绘制药-时曲线时每个时间点不少于5个数据为限计算所需的动物数量,尽量同一动物多次取样,一般雌雄各半。2)采样点:给药前需要采血作为空白对照,采样点的设计应兼顾药物的吸收相、平衡相、和消除相,以得到完整的药-时曲线,一般吸收相需要至少2-3个采样点,平衡相需要至少3个采样点,消除相需要至少4-6个采样点。整个采样时间应至少持续到3-5个半衰期或持续到血药峰浓度的1/20-1/10。为保证最佳采样点,可以在正试验之前进行预试验。3)根据测得的血药浓度随时间变化的数据(包括单次给药和多次给药)绘制药-时曲线,采用房室模型或非房室模型故断药代动力学参数,对于静脉给药应取得消除半衰期t1/2、表观分布容积Vd、血药浓度-时间曲线下面积(AUC)、清除率(CL)等;对于血管外给药,除上述参数外,还应取得峰浓度(Cmax)、达峰时间(Tmax)等。
药物吸收:对于经口给药,应尽可能同时进行血管内给药,提供绝对生物利用度。
药物分布:一般采用小鼠和大鼠,选择一个剂量(一般为有效剂量)给药后,以药-时曲线为参考,至少选择3个时间点(分别于吸收相、平衡相、消除相,每个点至少应有5个动物数据)测定药物在心、肝、脾、肺、肾、胃、肠道、生殖腺、脑、体脂、骨骼肌等组织的浓度。
代谢:药物在体内的代谢研究一般选用至少两种动物,一种啮齿类(一般选大鼠),一种非啮齿类(一般选犬)。在给药后分别采集血样、尿样和粪便,采用色谱方法分析其中可能存在的代谢产物,并确定代谢物的结构,研究药物在体内的转化类型、主要转化途径和可能设计的代谢酶等。
排泄:一般采用小鼠或大鼠,给药后按照一定的时间间隔分段收集尿、粪和胆汁的全部样品,每个时间点应至少有5个动物的数据,直至收集的样品测不到药物为止。测定收集样品中药物的浓度,记录药物排出的速度和排出量。
毒理学评价
临床前毒理学评价是通过各项毒理试验发现药物的毒性及其性质、中毒时间、靶器官、危害程度、解救措施等,同时推算临床试验的安全参考计量和安全范围的条件(中毒剂量、安全剂量范围),从而决定药物能否进入临床或确定临床中安全使用的条件等。主要包括急性毒性、长期毒性、特殊毒性等。
急性毒性试验:急性毒性试验是指动物单次或24h内多次给药后,一定时间内所产生的毒性反应。以了解新药急性毒性的强度,为长期毒性试验、特殊毒性试验、I期临床试验的剂量选择提供依据,同时也可以为临床毒副反应的监护提供参考。
试验设计:需符合一般动物试验的基本原则:随机、对照、重复。
试验对象:一般选用一种啮齿类动物和一种非啮齿类动物,需选择健康成年的动物,一般雌雄各半,动物数量根据动物种属和试验目的确定。
给药途径:一般至少包括临床拟用途径和一种能使药物较完全的进入循环的途径(如静脉注射)。
给药剂量:急性毒性试验重点在于观察动物出现的毒性反应,给药剂量应包括未见毒性反应到出现严重毒性反应的剂量,同时设置空白或溶剂(辅料)对照组。
观察时间和指标:给药后几小时内应严密观察,之后连续观察至少14天,观察的间隔和频率应适当以便观察毒性反应的出现时间、恢复时间和死亡时间等。观察指标包括动物外观、行为、分泌物、排泄物、死亡情况、体重变化等。
结果分析:分析不同剂量下药物产生的毒性、发生率、严重程度,判断每种反应的剂量-反应、时间-反应关系,可能涉及的组织、器官等。确定药物的无毒性反应剂量、严重毒性反应剂量,采用试验方法测定最大无毒性反应剂量(NOAEL)、最大耐受量(MTD)、最小致死剂量(MLD)等。也可以测定药物的半数致死剂量(LD50)。
长期毒性试验:长期毒性试验是反复多次给药与动物,观察药物产生的毒性反应。一般需连续给药14d以上,长期毒性试验可预测药物引起的不良反应的性质、程度、剂量-反应关系、时间-反应关系、可逆性、毒性靶器官等,其目的也是为临床试验给药剂量提供依据,降低临床试验受试者和上市后使用人群的用药风险。
试验对象:要求同急性毒性试验,通常选用大鼠和Beagle犬或猴,大鼠一般雌雄各10-30只,Beagle犬或猴为雌雄各3-6只。
给药方案:一般至少设置低、中、高三个剂量组和溶剂(辅料)对照组,其中高剂量组应使动物产生明显的毒性反应甚至出现个别动物死亡,低剂量应高于药效学试验的等效剂量并不使动物出现毒性反应。给药途径应与临床用药途径一致。一般应每天给药。
观察指标:长期毒性试验必须检测的指标有血液学指标(红细胞计数、血红蛋白等)、血液生化学指标(天冬氨酸氨基转换酶、丙氨酸氨基转换酶等)、尿液分析指标(外观、密度、pH值等)、脏器组织的病理学检查,此外也要根据药物的特点有针对性的增加相应的检测指标。
除上述毒性试验外,毒理评价还要考虑生殖毒性试验、遗传毒性试验、致癌试验等。
临床研究
由于人和动物的种属差异,候选药物在人体外或动物体内的疗效和毒性不能等同于在人体内的效应,甚至可能会因为体内过程差别而迥异;因此在临床前评价的基础上,还需要对候选药物在人体内的安全性和有效性做出评价,临床研究过程需要符合GCP的规范,确保研究结果准确可靠。根据其内容和目的可以分为I-IV期临床、生物等效性临床、桥接临床等。
生物等效性试验:是指用生物利用度研究的方法,一般以药代动力学参数为指标,比较同一种药物的相同或者不同剂型的制剂,在相同的试验条件下,其活性成份吸收程度和速度有无统计学差异的人体试验。
桥接试验:研究是指为原地域上市的药品能在新地域上市所开展的一系列研究,以评价该药物的种族敏感性,其可能是一项PK/PD试验,也可能是一项有效性试验,其目的在于利用已有的外部临床试验数据,外推该药品在新地域的相应适应症的有效性和安全性,缩短开发周期,降低开发成本,加速新药在新地域获准上市。
伞式研究vs篮式研究:伞式研究和篮式研究都是现代化临床设计的应用,二者均属于采用主方案的临床试验设计,以加速肿瘤药物和生物制品开发。篮式设计是指在同一个试验中,对不同的肿瘤病人采用同一种药物或药物组合进行治疗。伞式设计指在同一个试验中,对于同一种肿瘤的不同基因型采用不同的药物进行治疗。
非劣效vs优效临床试验:非劣和优效临床试验常用于阳性对照试验,评估试验药相对阳性对照药物的效果;一项临床试验在设计时即需明确是非劣/优效临床试验,并确定评价的界值和需要的样本量。在非劣效的临床设计中,得到非劣结果后,可以继续进行优效性检验,即非劣转优;但是优效性设计的试验没有得出优效性结论后不能接着进行非劣效检验。同理也有等效性试验。
I-IV期临床试验:对于创新药,要按照循序渐进的原则,从I期到IV期临床对其在人体内的安全性、有效性和药代动力学等进行系统的评价,这是新药研发过程最常见的临床试验。
图表:常见临床试验类型总结
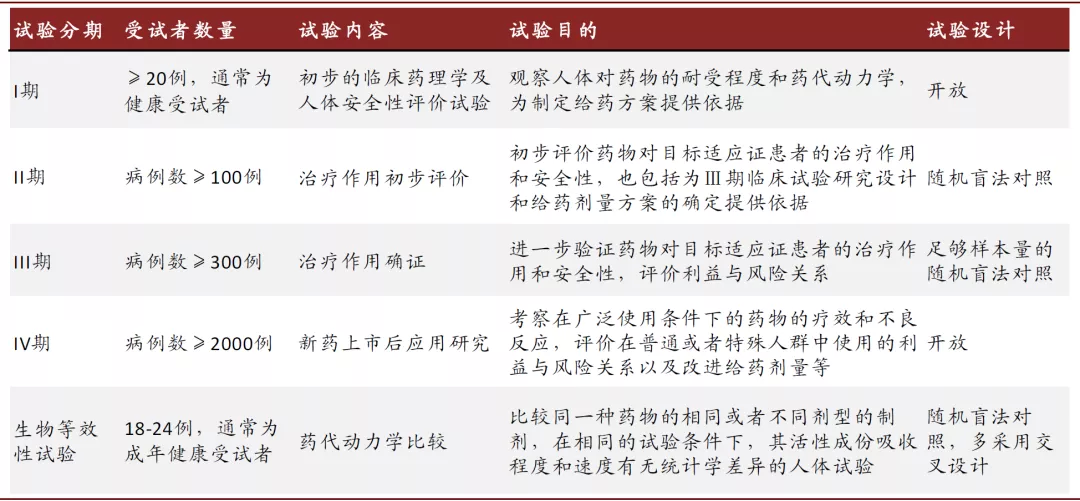
资料来源:药品注册管理办法,中金公司研究部
目标人群和患者选择:临床试验的主要目的之一是对药物用于患有特定疾病的人群中的疗效和安全性进行准确的评价,而临床试验的结果分析和推断都是在通过目标人群中选择一个代表性的样本进行的,因此患者的选择对该临床试验是否能达到试验的目的起着重要作用。目标患者人群通常是多样化的,因此需要设立一定的标准以降低其他因素(年龄、性别、机体功能状态、疾病严重程度等)对试验结果的影响,通常需要设立入选标准和排除标准,入选标准用来概括纳入研究的合适的目标患者人群,排除标准用来去除预期可对结果产生混杂影响的因素。
试验设计:在评价药物的安全性和有效性的临床试验中,平行分组设计是最常见的临床试验设计,该设计中每个受试者入组时随机接受一种治疗,直至临床试验结束。而在生物等效性试验中,常采用交叉设计,交叉设计是指每个受试者在不同阶段接受不同的治疗,每组患者按照不同的顺序接受不同的治疗,常见的有2×2交叉设计。在临床I期试验中,为了解药物的安全性和耐受性,通常采用剂量递增设计。
对照:临床试验中,多种因素会对试验产生偏移和变异,因此需要有良好的对照以为药物提供一个无偏、有效的安全性和有效性评价。对照组和试验组在人口学、临床等基线特征,以及治疗的评价、试验中变异可能产生的影响等方面都应该相似;基于此,才能认为试验组和对照组之间的临床结果差异是由于患者接收了不同的治疗这一单一因素产生的。按对照组接受的治疗可分为安慰剂、阳性(标准治疗)药物,也有不同剂量药物作为对照等。
随机化:在比较性临床试验中,通常使用随机化的方法将患者分配至各组,使每一个受试者都有同等的机会被分配到不同的组中,其目的不仅仅是确保不同组间具有可比性(除处理因素外其他基线特征一致),也是为临床试验结果的评估提供可靠的统计学检验方法。出于伦理学考虑,为了减少分入安慰剂组的患者比例,可以将不同数量的患者分配到治疗组和安慰剂组,如2:1等。有时为避免不同组的受试者的基线特征存在不均衡,也会采用分层随机的办法,先把样本按照某些影响结局的因素(年龄、性别、地理位置等)分层,在每一层中再进行随机化分配。为避免不同组之间受试者分配数量不均衡,也可以采用区组随机化方法。
设盲:在临床试验中,若受试者知道自己是在试验组或对照组,可能会刻意改变自己的行为或者寻求其他治疗。同样试验的研究人员如果知晓受试者的分组,可能也无法对试验组和对照组的研究对象做到“一视同仁”;为避免这些主观因素给试验结果带来偏倚,临床实践中通常采取设盲,单受试者不知道自己的分组情况为单盲,受试者和研究人员均不知道分组情况为双盲,受试者、研究人员、资料分析人员均不知道分组情况为三盲。
样本量:样本量的计算首先应考虑试验设计,包括对照的选择(安慰剂对照、阳性对照)、试验类型(优效性试验、非劣性试验、等效性试验等)、设计类型(平行设计、交叉设计等)、主要评价指标(定性、定量、生存时间等),其次明确统计分析方法做出效应量假定,然后根据试验要求的检验水准(α)、检验效能(1-β)、单双侧和试验组对照组分配比例;再根据相应的样本量估计方法计算所需样本量,最后根据试验脱落率、剔除率、依从性等做适当调整。
以常见的平行分组、安慰剂对照、有效性临床试验为例,假定评价指标是定量的,做均值t检验,标准差为σ,试验组和对照组的指标均值差异要达到Δ,试验组和对照组按照1:1分配,则每组所需样本量如下:

结果分析:临床过程中常常会发生预期之外的事情,如受试者可能因为各种原因在临床试验结束前退出试验,有的受试者甚至在随机化后没有任何资料,也有受试者中途转为其他的治疗方案等。因此哪些受试者纳入分析会对结果产生影响。ICH的指导原则中ITT原则(Intention to treat,意向治疗原则)是指根据受试者的意向治疗(即计划的治疗方案)可以比根据实际接受的治疗方案更好地评价疗效,即随机化后即纳入分析;但在实际中往往难以达到,因此常采用FAS(Full analysis set,全分析集),FAS是指由所有随机化的受试者中以最小的和合理的方法剔除受试者得出,未完成的数据观测值以最后一次观测值转接到最后的观测值(LOCF)。
评价指标的选择:临床试验评价指标要结合临床试验目的,根据指标是否容易量化、是否客观、是否可重复性等来做选择,通常包括主要临床终点和次要临床终点。在II/III期临床试验中通常用有效性指标作为主要临床终点,有效性指标通常应选取能够直接反映临床获益的指标,如肿瘤临床试验中通常选取总生存期作为主要临床终点。但有时反映临床获益的直接评价指标并不可行,如降压、降脂、降糖药物的临床获益通常是降低或延迟心脑血管事件的发生,但是其需要很长时间的观察,因此有时采用替代指标来评价药物的有效性,如血压降低值、血脂、血脂达标水平等;在肿瘤临床试验中,为了缩短观察时间,现也常用基于肿瘤评价的指标(如PFS、ORR等)作为主要临床终点。各个指标的含义在此不再赘述。
在生物等效性试验中,评价指标通常选取AUC和Cmax,一般要求AUC和Cmax的几何均值比的范围均在80-125%之间。
风险
新药研发失败,放量不及预期。
(编辑:张金亮)



 扫码下载智通APP
扫码下载智通APP